
Abstract
Polyploidy or whole-genome duplication (WGD) is a major event that drastically reshapes genome architecture and is often assumed to be causally associated with organismal innovations and radiations. The 2R hypothesis suggests that two WGD events (1R and 2R) occurred during early vertebrate evolution. However, the timing of the 2R event relative to the divergence of gnathostomes (jawed vertebrates) and cyclostomes (jawless hagfishes and lampreys) is unresolved and whether these WGD events underlie vertebrate phenotypic diversification remains elusive. Here we present the genome of the inshore hagfish, Eptatretus burgeri. Through comparative analysis with lamprey and gnathostome genomes, we reconstruct the early events in cyclostome genome evolution, leveraging insights into the ancestral vertebrate genome. Genome-wide synteny and phylogenetic analyses support a scenario in which 1R occurred in the vertebrate stem-lineage during the early Cambrian, and 2R occurred in the gnathostome stem-lineage, maximally in the late Cambrian–earliest Ordovician, after its divergence from cyclostomes. We find that the genome of stem-cyclostomes experienced an additional independent genome triplication. Functional genomic and morphospace analyses demonstrate that WGD events generally contribute to developmental evolution with similar changes in the regulatory genome of both vertebrate groups. However, appreciable morphological diversification occurred only in the gnathostome but not in the cyclostome lineage, calling into question the general expectation that WGDs lead to leaps of bodyplan complexity.
Similar content being viewed by others


Main
Polyploidy or whole-genome duplication (WGD) is a dramatic genomic event commonly invoked causally in organismal evolution1. The generally accepted ‘2R hypothesis’2,3 suggests that two rounds of WGD occurred during early vertebrate evolution (referred to as 1R and 2R); however, their timing and macroevolutionary consequences remain unclear4,5,6. Most studies agree that 1R occurred before the divergence of living vertebrates, but debate centres on whether 2R predated7,8 or postdated9,10,11,12 the divergence between cyclostomes and gnathostomes (Fig. 1c). Reconstruction of the ancestral vertebrate karyotype is fundamental to unravel the timing of 2R8,12,13,14,15, but this goal has been stymied by a dearth of cyclostome genomes. The recently described genome of the sea lamprey (Petromyzon marinus) has been interpreted to support 2R occurring before8 or after12 the gnathostome–cyclostome split, or not at all (with the karyotype diversity explained as the result of large-scale segmental duplications16,17). Analysis of the Arctic lamprey (Lethenteron camtschaticum) genome has suggested that 2R occurred in the gnathostome lineage while independent WGD event(s) might have occurred in the lamprey lineage11,18, perhaps shared with the hagfish11,19 (Fig. 1c). However, the lack of a hagfish genome assembly, the only major vertebrate group without a reference genome, has challenged attempts to constrain the number and phylogenetic timing of ploidy events in early vertebrate evolution. Here we describe the outcome of sequencing and comparative analysis of the genome of the inshore hagfish, Eptatretus burgeri (Fig. 1a,b).

a, Dorsal view of a young adult of the inshore hagfish E. burgeri, with the head to the top right. The teeth apparatus (and not a jaw) can be observed in a magnification of the head region of a fixed adult individual (a’). b, Fertilized egg of E. burgeri with a developing embryo at stage Dean 53 (ref. 48). Blood vessels can be observed from the exterior. c, Two competing hypotheses of vertebrate phylogeny. WGD events corresponding to the 2R hypothesis (lilac), to an alternative vertebrate 2R hypothesis (orange) and to those recently proposed in the lamprey lineage (light blue) are marked. Whether the lamprey-specific events actually occurred in a stem cyclostome remains elusive. d, Hi-C contact heatmap of the corrected hagfish genome assembly ordered by cluster (chromosome) length. Dashed boxes indicate the cluster boundaries. e, Completeness assessment of the genome assembly of the inshore hagfish E. burgeri genome (red), three lamprey species (blue) and two jawed vertebrates (green). Number of conserved metazoan orthologues (metazoa_odb10 dataset, containing 954 BUSCOs) is indicated for each case. F. E., Far Eastern. f, Correspondence analysis (CoA) on RSCU values was performed using the nucleotide sequences of all predicted genes concatenated for individual species. The percentage of variance is indicated for each axis. g, CoA of amino acid composition, with the percentage of variance indicated for each axis. In f and g: red, hagfish; blue, lamprey; green, jawed vertebrates; black, invertebrates.
Chromosome-scale assembly and genome annotation
Similar to the lamprey20, the hagfish genome undergoes somatic programmed DNA rearrangement in the way of chromosome elimination21, making it crucial to obtain a reference assembly from a germline source. We sequenced DNA extracted from the testis of a single sexually mature male of E. burgeri and generated a preliminary draft assembly using ~240X of short-read Illumina data assisted by a Chicago in vitro proximity ligation assay at Dovetail Genomics22 (Supplementary Table 5). We estimated the genome of E. burgeri at 3.12 Gb on the basis of k-mer frequency distribution (Extended Data Fig. 1a and Supplementary Table 4) in line with other hagfish species (~2.2–4.5 Gb)23. Chromosome conformation capture (Hi-C) data obtained from the testis DNA of a second individual were used to further scaffold the genome into a final assembly (v.4.0) containing 19 contact clusters, which we consider as chromosomes for subsequent analyses (Fig. 1d), and 9,295 unplaced scaffolds and contigs (Methods and Supplementary Tables 6–10). The genome was annotated following the Ensembl annotation pipeline24, assisted by RNA-seq from 9 different adult tissues, and previous embryonic and juvenile transcriptomics data19 (Supplementary Table 11 and Methods). We generated a final gene dataset of 16,513 protein-coding genes (with 27,960 transcripts), 446 long intergenic non-coding (linc)RNAs and a minority of other classes of non-coding RNA genes (Extended Data Fig. 1b). A total of 180 microRNA (miRNA) genes were found in the E. burgeri genome conserved with the hagfish Myxine glutinosa25 belonging to 77 miRNA families and catalogued at MirGeneDB.org26. The germline haploid number of E. burgeri is 26. However, chromosome elimination occurs in somatic tissues of the hagfish, by which, in the case of E. burgeri, 8 pairs of microchromosomes are eliminated during development21 (somatic n = 18). Cluster 19 and unplaced contigs/scaffolds probably correspond to these difficult-to-assemble microchromosomes, which presumably consist mainly of highly repetitive sequences and contain almost no protein-coding genes21. Consistently, 98.3% (16,240/16,513) of annotated genes are located in clusters 1–18.
BUSCO analyses show high levels of completeness of the hagfish genome (96.0 and 94.2% of single orthologues are present in the assembly and annotation, respectively; Fig. 1e and Extended Data Fig. 1c). GC-content distribution pattern analysis of the hagfish and other chordate genomes shows that the E. burgeri genome represents an intermediate condition between the lamprey and other chordates (Extended Data Fig. 1d), although having an overall GC content similar to that of the lamprey (46.7% and 48.1% for the hagfish and lamprey, respectively). While lamprey protein-coding gene sequences have been demonstrated to pose difficult challenges for comparative analyses due to their high GC content27 (64.0%), the lower content in hagfish coding sequences (50.4%) is within the typical range of most gnathostomes and non-vertebrate chordates (42.5%–53.4%; Extended Data Fig. 1e,f and Supplementary Table 12). Lamprey represents an outlier in terms of both codon usage bias and amino acid composition, while the hagfish is more similar to other vertebrates (Fig. 1f,g). The hagfish genome contains, on average, significantly longer introns and intergenic regions than other vertebrates (P < 2.2 × 10−16, two-sided Wilcoxon rank-sum test), while the average length of coding sequences is similar to that of other chordates (Extended Data Fig. 1g, Supplementary Fig. 4 and Supplementary Tables 18–22). This might explain why hagfish genomes are larger than lamprey genomes23. Altogether, the hagfish genome provides essential, complementary information to lamprey genomes, especially in analyses such as gene tree reconstruction and comparative genomics.
Hagfish phylogenomics and gene family evolution
Whether hagfish form a clade with lampreys (Cyclostomata) or represent the sister group to all other vertebrates (including lampreys) has depended on whether molecular or morphological evidence are considered (Fig. 1c). Morphological studies historically supported cyclostome paraphyly but more recent analyses have recovered cyclostome monophyly (reviewed in ref. 28). Phylogenies inferred from molecular evidence have almost exclusively recovered cyclostome monophyly (reviewed in ref. 25). We used Bayesian inference to reconstruct the phylogeny of vertebrates from an alignment of 190 single-copy genes in all taxa analysed (84,017 sites), strongly supporting a monophyletic Cyclostomata (Fig. 2 and Extended Data Fig. 2a). We calculated the likelihood of gene duplication and loss patterns under the competing phylogenetic hypotheses29 (see Methods), finding that patterns of gene gains and losses better fit cyclostome monophyly. To further compare the two alternative hypotheses of hagfish relationships, an approximately unbiased (AU) test30 was performed, which strongly rejected cyclostome paraphyly (log likelihood difference = 7,947.7, AU = 0.004, multiscale bootstrap probability < 0.001). These results corroborate previous molecular analyses and recent morphological studies28, supporting the view that cyclostomes are monophyletic.

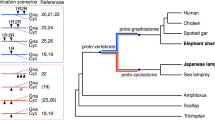
Time-calibrated rooted phylogeny of vertebrates and two non-vertebrate species with 95% credibility intervals for clade divergence times indicated by red bars on nodes. The phylogenetic tree was obtained with Bayesian inference (Extended Data Fig. 2a) and all nodes were recovered with a posterior probability of 1. Numbers of gene family gains (green, novel homology group (HG); blue, novel core HG) and losses (red) are indicated in selected nodes (see text). Dated WGD events, including 1R, 2R and cyclostome-specific event (CR) described in this study, are indicated with coloured rectangles. The hagfish position is highlighted with a thickened line and bold font. Geological periods are colour-coded at the bottom: Ediac., Ediacaran; Cambr., Cambrian; Ordo., Ordovician; Sil., Silurian; Devon., Devonian; Carbonif., Carboniferous; Perm., Permian; Palg., Paleogene; Ng., Neogene. Animal illustrations kindly provided by Tamara de Dios Fernández; human, zebrafish, lamprey and hagfish illustrations reproduced with permission from ref. 133.
To better understand the genomic changes accompanying major transitions in chordate evolution, we used a phylogeny-aware comparative genomic approach31,32,33 to infer ancestral gene complements and gene family gains and losses across the vertebrate tree (Fig. 2, Extended Data Fig. 3 and Supplementary Tables 23–26). We observed two peaks of gene novelty in both the vertebrate and gnathostome stem-lineages (novel genes: +560 and +771, respectively), also characterized by a very low amount of gene losses (−341 and −382, respectively) when compared with other deuterostome and chordate nodes (Fig. 2 and Extended Data Fig. 3a). Furthermore, the fraction of highly retained novel gene families (also known as novel core genes, that is, genes that are not lost in descendant lineages and by convention indicated by ++) is the highest in the last common ancestors of vertebrates, gnathostomes and cyclostomes (novel core genes: ++81, ++86 and ++98, respectively; Fig. 2 and Extended Data Fig. 3a). These are notably larger than those observed in other major evolutionary episodes in metazoan evolution31,33, but generally similar to a recent study using more chondrichthyan but only two invertebrate genomes34, suggesting that the emergence of new gene families played important roles in the origin and diversification of early vertebrates. Gene Ontology (GO) enrichment analyses demonstrate that the origin of vertebrates was characterized by the appearance of genes involved in signalling pathways, cell communication and transcriptional regulation (Supplementary Tables 24 and 25), while novel core genes involved in immunity played an important role in the origin of gnathostomes (Supplementary Tables 24 and 26). Consistently, gnathostomes and cyclostomes convergently evolved independent adaptive immune systems, based on immunoglobulins in the former, and in variable lymphocyte receptors in cyclostomes35 (Supplementary Fig. 6 and Supplementary Table 27). The largest fraction of gene losses occurred in the ancestral cyclostome lineage (Fig. 2), suggesting that a strong asymmetric reduction of gene complements accompanied the early evolution of the group. For instance, the hagfish genome lacks several vision and circadian rhythm-related genes, probably associated with its vestigial eyes (Supplementary Fig. 7 and Supplementary Table 28). Inferred rates of gene duplication (irrespective of the duplication mechanism) across Metazoa identify widespread duplications associated with the vertebrate and teleost stem-lineages (Extended Data Fig. 2b,c), probably reflecting the 1R, 2R and teleost 3R WGD events36. We also inferred high duplication rates in each of the lineages leading towards crown-gnathostomes and lampreys (Extended Data Fig. 2b,c) which might suggest large-scale duplications associated with these groups, consistent with the WGD events proposed recently12,18. This type of analysis, however, cannot discriminate between WGD and other large-scale gene duplication mechanisms.
Conserved Hox cluster evolution in cyclostomes
The number of Hox clusters and ancestral WGD events are usually correlated; hence, the former has been used as a genomic marker of the latter. The presence of 6 Hox clusters in lamprey genomes17,18 has been interpreted to indicate the possibility that more than two WGD events occurred in this lineage18. We have extended previous observations in E. burgeri19 to confirm the presence of 40 Hox genes arranged in 6 complete Hox clusters (Fig. 3). Two of the hagfish clusters are located in the same chromosome (cluster 3), separated by >80 Mb, probably the result of chromosomal shuffling due to the intense reorganization of the hagfish genome from ancestral chromosomes (ACs; see below). Phylogenetic analyses of Hox coding sequences have long proven inconclusive to determine the orthology relationship between lamprey and hagfish Hox counterparts19. We thus applied a microsynteny conservation approach using extended Hox loci which, together with phylogenetic analyses of selected non-Hox syntenic genes, allowed us to establish clear one-to-one orthologous correspondences between hagfish and lamprey Hox clusters, named α to ζ after the lamprey clusters18 (Fig. 3, Extended Data Fig. 4a–d, Supplementary Fig. 8 and Supplementary Tables 29 and 30). This suggests that the crown-cyclostome already possessed 6 Hox clusters, distinct from the ancestral crown-gnathostome, which possessed 4 clusters (Supplementary Fig. 9). This observation provides further evidence of cyclostome monophyly, by suggesting that lampreys and hagfish share a genome history exclusive of gnathostomes. This implies that the events suggested from the different analyses of the Arctic lamprey genome, two extra WGDs18 or a triplication11, might have occurred in early cyclostome evolution, probably before the lamprey and hagfish divergence11,19.

Schematic representations of Hox clusters and syntenic genes of the inshore hagfish (E. burgeri, middle), the sea lamprey (P. marinus, bottom) and a reconstruction of the complement of the last common ancestor of hagfishes and lampreys (top). Genes are represented by colour-coded arrows whose direction marks the sense of transcription: Hox genes in red, non-Hox genes coloured by homology (legend at right). The block between Evx-ζ and Creb2/5/7-ζ is assembled downstream of the Hox-ζ cluster, separated by 7 genes. This might be a misassembly, and their ‘natural’ upstream position is marked by a dashed line. The sea lamprey genome scaffolds and hagfish Hi-C clusters in which Hox clusters are located are indicated to the right of each cluster, with cluster 3 separated into 3L (0–107.78 Mb) and 3R (107.78–194 Mb). Black asterisks mark genes placed at opposite sides of a cluster in the hagfish and the lamprey, but placed at one side of the ancestrally reconstructed cluster on the basis of comparisons with gnathostomes (Supplementary Fig. 9); hashes denote genes present in lampreys in the same chromosome but at a long distance (see Supplementary Fig. 8); white asterisk denotes Nfe2-ζ inferred due to its presence in the Arctic lamprey. Animal illustrations kindly provided by Tamara de Dios Fernández; lamprey and hagfish illustrations reproduced with permission from ref. 133.
Ancestral vertebrate karyotype
The reconstruction of the pre-WGD vertebrate proto-karyotype by means of macrosynteny analysis stands as the most robust approach to test the 2R event and its phylogenetic position37. Earlier attempts at reconstructing the ancestral vertebrate karyotype have yielded widely disparate outcomes, indicating 10–13 (refs. 14,16) or 17–18 (refs. 8,11,12,13,38) ancestral pre-duplicative chromosomes. These reconstructions have also unveiled a perplexing scenario where lampreys’ divergence from gnathostomes occurred either before or following the 2R event8,11,12,18. To shed new light on early vertebrate genome evolution, we performed a macrosynteny conservation analysis between gnathostomes, cyclostomes and selected invertebrate deuterostomes. First, to minimize noise from lineage-specific fusion and fission events, we reconstructed ancestral chicken39 and spotted gar40 genomes using elephant shark41 as an outgroup, obtaining an almost perfect one-to-one chromosome orthology (Supplementary Table 31). Next, to infer the ancestral vertebrate karyotype, we elaborated a map of homology relationships between the genes of these slow-evolving gnathostome genomes with the chromosome-level genome assembly of the sea cucumber Apostichopus japonicus (echinoderm) as a pre-duplicative outgroup species42. With this, we inferred a proto-vertebrate karyotype of 17 ACs (Supplementary Fig. 15 and Supplementary Table 32).
We mapped genes from the Belcher’s lancelet (Branchiostoma belcheri) genome43 to each AC (Methods and Supplementary Fig. 21) using very stringent criteria, requiring homology relationships of an amphioxus gene with both a sea cucumber gene and several chicken and/or spotted gar genes, and all anchored to the same AC. In total, we mapped 5,065 Belcher’s lancelet genes to AC1–17 (ranging from 115 to 534 genes in AC1–AC16; AC17 consisted of only 20 genes and was thus excluded from several subsequent analyses; Supplementary Table 34). With these in hand, we corroborated our ancestral vertebrate karyotype reconstruction through comparisons with the chromosome-scale genome of the amphioxus Branchiostoma floridae12 (Supplementary Tables 35 and 36).
Our inference of an ancestral karyotype with 17 ACs matches a previous study8, has minor differences with other 17-chromosome inferences12,13 and depicts one less chromosome than more recent studies11,38 (Supplementary Tables 37 and 38). In our model, 4 of the 17 ACs (AC1, AC2, AC3 and AC6) each correspond to 2 or 3 linkage groups (putative chromosomes) of the sea cucumber genome (Supplementary Fig. 15 and Supplementary Table 32) as well as to distinct homologous chromosomes in B. floridae (Supplementary Table 36), suggesting that these 4 ACs probably originated via fusions of ancestral chordate chromosomes in the vertebrate lineage before 1R38. The difference from previous 18-chromosome models is that while we consider that the vertebrate AC3 is a single chromosome resulting from a pre-1R fusion event of two ancestral chordate chromosomes (Supplementary Fig. 23a), others11,12,38 consider that these two chromosomes remained separate through 1R (Nakatani’s Pvc8 and 9, or Simakov’s CLGQ and CLGI, respectively). While Pvc8/CLGQ and Pvc9/CLGI are consistently co-located in gnathostome chromosomes, they remain separate in invertebrate karyotypes11,12,38. We did not find any signals of linkage between Pvc8/CLGQ and Pvc9/CLGI in the lamprey and the hagfish genomes (Supplementary Figs. 25 and 26). Therefore, there exist two alternative scenarios: (1) the 18-chromosome model implies that two independent pairwise fusions occurred after 1R in a stem gnathostome, mimicking a single pre-1R fusion event (Supplementary Fig. 23a); and (2) our 17-chromosome model requires symmetric fissions of two AC3-derived post-1R chromosomes occurring in an ancestral cyclostome (Supplementary Fig. 23b). Although in silico simulations show that a scenario of pairwise post-1R fusions would not be extremely rare (30% of cases expected by chance; Supplementary Table 40 and Methods), we believe the pairwise fissions to be more plausible given the higher level of reorganization found in cyclostome karyotypes (see next section). Altogether, while we propose a scenario involving 17 ancestral vertebrate chromosomes, a scenario with 18 chromosomes11,38 is also possible.
Importantly, all ACs correspond to sets of four (11/17) or three (6/17) paralogous chromosomes in the gnathostomes chicken and gar, a strong genome-wide pattern consistent with 2R2,11,12,13. We stringently selected 701 sets of orthologous genes (Methods and Supplementary Table 39) between the sea cucumber, chicken, spotted gar and an AC gene (from B. belcheri), and built robust chromosome-level phylogenies with a median of 38 concatenated gene sets across each of the ACs (Extended Data Fig. 5a and Supplementary Table 39). The highly supported, clear-cut topologies further support the existence of 2R in gnathostomes and depict the exact evolutionary trajectory from each AC to their modern chicken and spotted gar descendants (Extended Data Fig. 5a,b). Our reconstruction of gnathostome karyotype evolution involved 8 fusion events that took place after 1R but before 2R (Extended Data Fig. 5b), similarly to what has been previously found by others8,11,12 (Supplementary Table 41). Furthermore, we found a significant gene retention asymmetry after 2R, with a median of 1:2.28 genes per ohnologous (duplicates that originate through WGD, after ref. 3) chromosome pair, but not after 1R (median 1.16; P = 3.4 × 10−7, Wilcoxon rank-sum test; Extended Data Fig. 5b). This pattern is consistent with previous studies suggesting that 1R was an autotetraploidization event and 2R an allotetraploidization event11,12 (but see ref. 44 on asymmetric gene retention after teleost 3R autotetraploidy).
Gnathostomes and cyclostomes share 1R but not 2R
We next tested hypotheses of WGD timing relative to cyclostome divergence. We assessed the phylogenetic signal of hagfish and lamprey genes anchored to 661 orthologous gene sets (Supplementary Table 42), including elephant shark orthologues as a control for the 2R signal and amphioxus genes as outgroups. Approximately 73.2%, 79.1% and 75.7% of trees including hagfish, lamprey or both hagfish and lamprey orthologous genes, respectively, are compatible with shared 1R (Fig. 4a). However, while 99.5% of elephant shark gene tree topologies are 2R-compatible, only 19.1% of hagfish, 10.6% of lamprey and 8.2% of cyclostome (including both lamprey and hagfish) gene trees are compatible with a 2R history (Fig. 4a and Supplementary Files 5–8). Thus, we find strong support only for 1R as shared among cyclostomes and gnathostomes.

a, Phylogenetic support of gnathostome and cyclostome genes for 1R and 2R. Elephant shark, hagfish, lamprey or both cyclostomes’ genes (both hagfish and lamprey genes included) were analysed as test genes in the context of spotted gar and chicken gene phylogenies by each AC (using amphioxus genes) and orthologous sea cucumber genes (outgroup). Left: possible positions where test genes can branch, supporting or not 1R or/and 2R (see legend). Middle and right: statistics of supporting (blue) or not supporting (orange) gene phylogenies from each species’ tested genes. All phylogenetic trees are available in Supplementary Files 5–8. b, Formula to calculate the OR between two chromosomes. Dark cyan denotes genes from the AC, retained in modern chromosomes; white indicates gene loss. c, OR values distribution between WGD-generated paralogous (ohnologous) chromosomes in chicken (top left) and spotted gar (top right), and the artificially split chromosomes in chicken (bottom left) and spotted gar (bottom right). Dashed lines mark OR = 0.15. d, OR values distribution between putative ohnologous chromosomes in hagfish (top left) and lamprey (top right), and the artificially split chromosomes in hagfish (bottom left) and lamprey (bottom right). e, OR values distribution between chicken and spotted gar (top) and between hagfish and lamprey (bottom) orthologous chromosomes. f, Numbers of mutually ohnologous chromosomes in cyclostome genomes that correspond to each one of the 16 reconstructed ACs. g, Retention profile clustering analysis of cyclostome chromosomes deriving from AC2. Retained genes are denoted by dark cyan lines. Five putative orthologous chromosome pairs are defined. Note that AC17 was excluded from the analyses depicted in c–f because of the low number of genes we recovered (20 genes). Animal illustrations kindly provided by Tamara de Dios Fernández; chicken, spotted gar, lamprey and hagfish illustrations reproduced with permission from ref. 133.
To further confirm the timing of the 1R event, we investigated whether signals of the inferred four pre-1R and eight post-1R fusion events are present in cyclostomes. When assessing how hagfish and lamprey chromosomes descended from the 17 ACs, we found that the hagfish genome displays a large amount of rearrangement (at least 52 fusions detected), making any signal of hypothetically shared events unreliable (Extended Data Fig. 6b). However, most lamprey chromosomes are descendants of single ACs12 (Extended Data Fig. 6a), making the lamprey a better model to investigate these rare genomic changes. We found that the sea lamprey genome17,45 bears signals of three (AC1, AC2 and AC6) and the hagfish genome of two (AC1 and AC2) of the four pre-1R fusions (Supplementary Figs. 25 and 26). On the other hand, similar to previous studies11,12 we did not find any reliable signal of the eight post-1R fusions detected in gnathostomes, suggesting that the lamprey and hagfish diverged after the 1R but before all eight post-1R/pre-2R fusions11,12. Taken together, our comprehensive phylogenetic analysis and the constraints given by pre- and post-1R chromosomal fusions provide strong evidence in favour of a pan-vertebrate 1R event, but constrains 2R to the gnathostome lineage as recently suggested in similar analyses11,12.
Cyclostome-specific whole-genome triplication
It has been suggested that the lamprey genome has been shaped by either three duplicative events18 or a hexaploidization11. The presence of six orthologous Hox clusters in both the lamprey and the hagfish (Fig. 3) implies that this is the ancestral condition for cyclostomes and supports the triplication event11,19. Although we find that multiple chromosomes and large chromosomal sections are descendant copies of each AC in both cyclostome groups, the extensive rearrangements observed in the hagfish and the large haploid number in the lamprey impede chromosome-level macrosynteny conservation analysis to distinguish intraspecific ohnologous and interspecific orthologous relationships. To confidently infer karyotype evolution in cyclostomes, we developed a new metric, the ‘overlapping ratio’ (OR), to measure the similarity of gene retention profiles of any two chromosomes hypothetically descending from a common AC (Fig. 4b and Supplementary Fig. 29; gene-poor AC17 was excluded from this analysis, which required at least 20 genes retained in each descendant chromosome). A retention profile is defined by a vector listing the presence or absence of genes on a modern vertebrate chromosome from their corresponding AC. Therefore, we expect the OR of chromosomes deriving from a duplication event to be significantly higher than that of chromosomes deriving from an ancestral fission followed by gene translocations. As proof of concept, we applied this metric to gnathostomes: knowing that their genomes have been shaped by the 2R event, we found that the median OR of ohnologous chromosome pairs in chicken or spotted gar was 0.49 (interquartile range, IQR: 0.44–0.56) and 0.54 (IQR: 0.47–0.65), respectively (Fig. 4c), while OR value for simulated fission-derived chromosome pairs was never larger than 0.15, indicating that ohnologous chromosomes indeed share more retained genes (Fig. 4c and Methods).
We then applied the OR metric to the sea lamprey (after correcting misassemblies, assisted by a meiotic map of the Pacific lamprey Entosphenus tridentatus17 and confirmed by the recent chromosome-level genome assembly45) (Supplementary Table 50 and Supplementary Fig. 24) and the hagfish, defining ohnologous chromosome pairs as those with OR > 0.15. We found that the median OR between putative ohnologous chromosomes was 0.30 (IQR: 0.23–0.36) and 0.29 (IQR: 0.23–0.37) for the lamprey and hagfish, respectively (Fig. 4d, Extended Data Fig. 7a,b and Supplementary File 9). Using this approach, we found that most ACs analysed (12/16 or 75%) have descended into three or more mutually ohnologous chromosomes in both the lamprey and hagfish (Fig. 4f), suggesting that at least a second WGD might have occurred in cyclostomes. In both genomes, at least five chromosomal regions are direct descendants of each of the same five ACs (1, 2, 6, 10 and 14), with 3 ACs contributing to 6 chromosomes each in the lamprey (Fig. 4f). We do not find more than 6 descendant copies from any AC, supporting a whole-genome triplication in the cyclostome lineage as previously proposed in the analysis of the lamprey genome11. The distribution of multiplicity across ACs is highly correlated across the two species (Spearman ρ = 0.91), suggesting that this triplication event is conserved between the lamprey and hagfish and thus occurred in an ancestral cyclostome. It is expected that OR will decrease with each WGD (Supplementary Fig. 29), hence the lower value in cyclostomes is consistent with the occurrence of this larger polyploidy event. While our data do not definitively rule out the possibility of two cyclostome-specific WGD events followed by extensive chromosome losses, this scenario is less plausible than a single triplication event, particularly given the absence of instances with eight copies of any chromosomal region in the lamprey or the hagfish.
To further confirm that this proposed triplication event is shared by hagfish and lamprey, we extended the use of the OR to detect putative orthologous chromosomes. During the process of diploidization after a polyploidy, two descendant chromosomes diverge and fix their mutations independently, hence it is expected that interspecific orthologous chromosomes will have more similar gene retention profiles than intraspecific ohnologous chromosomes (as long as rediploidization precedes speciation46). Accordingly, orthologous chromosomes of chicken and spotted gar have a median OR = 0.96 (IQR 0.95–0.98; Fig. 4e and Supplementary Table 44) and clustering-based analysis based on gene retention profiles places chicken and gar orthologous chromosomes closer to each other, completely reflecting the phylogenetic signal (Extended Data Fig. 7c and Supplementary File 10). When we applied this approach to cyclostome genomes, we found the median OR = 0.84 (IQR: 0.74–0.91; Fig. 4e and Supplementary Table 45) for 52 (~87%) chromosome pairings between lamprey and hagfish that putatively represent 1:1 orthologues (higher than that of ohnologous chromosomes; Supplementary Table 45) and only 8 (~13%) one-to-two or two-to-one ambiguous relationships, probably due to secondary independent chromosome losses in either group. Clustering analysis of retention profiles recovers orthologous relationships between lamprey and hagfish (Fig. 4g and Supplementary File 11). Overall, intra- and interspecific gene retention profile analyses indicate that a triplication event took place in the cyclostome stem-lineage; we refer to this as CR, to avoid confusion with the gnathostome-specific 2R event.
Increase of developmental regulatory complexity
To investigate the immediate consequences of the independent CR event on cyclostome genome evolution, we first asked whether retained duplicates (ohnologues) are especially associated with developmental functions in the hagfish as in their gnathostome counterparts13,47. GO enrichment analysis shows that hagfish gene ohnologues are also significantly enriched for functions associated with developmental processes (Extended Data Fig. 8a,b). Gnathostomes have increased their regulatory complexity (higher number of regulatory regions per gene), particularly of developmental ohnologues47. We identified accessible chromatin regions (ACRs) as putative non-coding regulatory elements in the hagfish genome with an assay for transposase-accessible chromatin coupled to sequencing (ATAC-seq), using a total of two embryos of E. burgeri, each at a different stage48 (45 and 53; Supplementary Fig. 32). We found a significantly higher number of ACRs per gene than in the cephalochordate amphioxus, similar to what has been observed in gnathostomes47 (Fig. 5a), particularly in distal regions from transcriptional start sites (Fig. 5b,c and Extended Data Fig. 8c). This pattern is especially evident in developmental genes (Fig. 5d and Extended Data Fig. 8d–f), implying that their higher retention after a cyclostome CR event is underlain by a more complex regulatory landscape of developmental genes, as in gnathostomes47.

a, Distributions of the ACR numbers within the cis-regulatory regions of each gene (see Methods). n = 28,497 (amphioxus), n = 23,183 (zebrafish), n = 22,184 (medaka), n = 15,213 (chicken), n = 23,256 (mouse) and n = 16,951 (hagfish) genes. ***P < 2.2 × 10−16, Bonferroni-adjusted, two-sided Wilcoxon rank-sum tests. b, Numbers and fractions of ACRs with respect to genomic annotations in each species. Promoters, between 1 kb upstream and 0.5 kb downstream of annotated transcription start sites (TSSs); proximal, within 5 kb upstream and 1 kb downstream of annotated TSSs, but not overlapping promoters; exonic, within exons of protein-coding genes but not overlapping proximal regions; distal, not in aforementioned locations. c, Cumulative proportion of the distance of ACRs from the closest TSSs in each species. For the result with scaling based on the average length of intergenic regions of each species genome, see Extended Data Fig. 8c. d, The distribution of ACR numbers across different classes of genes, according to PANTHER Gene Ontology database (devel., developmental ohnologues; non-dev., non-developmental ohnologues; non-ohnol., singletons). n = 143 (devel. ohnol.), n = 816 (non-devel. ohnol.) and n = 7,303 (non-ohnol.) genes. P values from Bonferroni-adjusted two-sided Wilcoxon rank-sum tests are indicated. e, Distribution of fates of ohnologous families after WGD. Red., potential redundancy; Subf., potential subfunctionalization; Spec., potential specialization. f, Number of ohnologues with strong specialization expressed in hagfish tissues. In a and d, boxes correspond to the median (centre line) and the first and third quartiles. Whiskers extend to the last point no further than 1.5× the interquartile range from the first and third quartiles. For a–d, see Supplementary Tables 52–57 for detailed statistical information, including P value for each pairwise comparison. Animal illustrations kindly provided by Tamara de Dios Fernández; chicken and hagfish illustrations reproduced with permission from ref. 133.
In gnathostomes, retained duplicates can evolve via expressional specialization (reduction of expression domains of one of the ohnologues)47, probably coupled to neofunctionalization rather than subfunctionalization (differential erosion of enhancers)49. Taking advantage of adult transcriptome data across nine organs (see Methods), we next analysed the putative fates of hagfish ohnologues after CR (Supplementary Fig. 31). Hagfish duplicates also tend to reduce their expressional domains: over 68% and 71% of gene families subfunctionalized or specialized in the hagfish and chicken, respectively (Fig. 5e). Hagfish ohnologues that have potentially restricted their expression domains (subfunctionalization or specialization) are associated with a larger amount of regulatory elements and a higher sequence evolutionary rate than those that have maintained the ancestral patterns (Extended Data Fig. 8g,h), similar to gnathostomes (Extended Data Fig. 8i)47. Furthermore, the largest portion of ohnologues with strong specialization (one or two ancestral expression domains) are expressed in the brain (Fig. 5f), mirroring the pattern observed in gnathostomes47 (Extended Data Fig. 8j). In summary, our results indicate that cyclostomes and gnathostomes followed parallel evolutionary pathways after their independent WGD events. Genes gained a larger regulatory complexity, mostly on distal regions and especially in duplicates with developmental functions, which tend to be retained more often. Furthermore, specialization is a common fate of ohnologues associated with faster sequence evolution and the acquisition of novel regulatory elements that drive their tissue-specific expression. Alternatively, the possibility that a decrease in the number of regulatory elements took place in the amphioxus cannot be confidently ruled out.
Impact of WGD events on vertebrate morphological diversity
Hypotheses on the role of WGD events in the origin and elaboration of the vertebrate bodyplan range from deterministic to permissive4. There can be no doubt that many vertebrate and gnathostome novelties are contingent on gene paralogues that are the product of the 1R and 2R events, although whether WGD played a causal role remains unclear. We employed two tests of a causal relationship: (1) absolute timing of the WGD events and the clades with which they are causally associated and (2) contrast in morphological phenotypic diversity before and after the WGD events. Using a dataset of 177 genes and 33 fossil calibrations, we provide estimated times at which duplicated subgenomes diverged. We adopted a sequential Bayesian approach in which the posterior clade age estimates from our species timescale were used as prior on the speciation nodes in our concatenated gene tree; to achieve statistical consistency, this requires that the same molecular loci are not used in the two analyses50. A parallel analysis in which the concatenated gene tree was calibrated using the original fossil calibrations yielded results that are less precise but otherwise not materially different. Interpretation of the results depends on the nature of the ploidy event; in the case of autopolyploidy, we estimate the minimum timing of rediploidization, when the two subgenomes derived from WGD stopped homologously recombining, a process that can be asynchronous and span dozens of millions of years44,46,51,52,53,54. In the case of allopolyploidy, the age estimate represents the speciation event that isolated each of the 2 subgenomes that later came together to form an allopolyploid genome55. Our relaxed molecular clock analyses estimate the 1R event to have occurred 535.3–524.8 Ma (early Cambrian), 14.3–29.2 Myr before the divergence of crown-vertebrates (510.5–506.1 Ma; middle Cambrian) (Fig. 2); the CR event is dated to 500–492 Ma (late Cambrian), 23.5–36.5 Myr before the divergence of crown-cyclostomes (468.5–463.5 Ma; Middle Ordovician) (Fig. 2); and the 2R event is dated to 498.4–485.2 Ma (late Cambrian–earliest Ordovician), 35.1–53.3 Myr before the divergence of crown-gnathostomes (450.1–445.1 Ma; Late Ordovician) (Fig. 2).
To characterize morphological disparity across WGD events, we compiled a phenotype matrix composed of 577 traits for 278 living and fossil chordates encompassing all aspects of morphology, which we subjected to pairwise distance analysis followed by ordination using non-metric multidimensional scaling (NMDS) (Fig. 6a). This multivariate approach groups organisms with similar suites of characteristics while separating organisms with dissimilar traits, providing us with a relative measure of bodyplan diversity. The results of this analysis show that each genome duplication is followed by an increase in morphological disparity through occupation of novel regions of morphospace (Fig. 6b), but the majority of chordate disparity (88–97% of the morphospace encompassed by a vertebrate convex hull) emerged subsequent to the 2R event (Fig. 6b). Thus, while 2R and CR are of comparable antiquity, there is a stark contrast in terms of bodyplan evolution and species diversity between the descendants of 2R and the other WGD events.

a,b, Morphological disparity across vertebrates. Non-metric ordinations are presented, highlighting the morphological variance among (a) taxonomic lineages of extant and extinct vertebrates and (b) the descendants of 3 whole-genome duplication events. Convex hulls have been fitted around groups. The underlying tree was derived from a consensus of relationships from the literature.
Discussion
The sequencing of a hagfish genome has enabled us to better understand the evolution of early vertebrates. First, our analysis of vertebrate genomes, including the hagfish, now establishes a robust and accurate history of WGD events in early vertebrates, corroborating the idea that cyclostomes diverged from gnathostomes after the 1R but before the 2R event. This is consistent with early9,10,13 and recent studies on the matter that included the lamprey11,12. We think that debate over the timing of 2R5 can now be concluded. Our hagfish genome also confirms an additional genome-wide duplicative event in stem-cyclostomes, CR, which most probably was a triplication, as previously suggested11. Thus, key vertebrate innovations (for example, elaborate tripartite brain, neural crest cell-derived tissues among other novelties56) originated in a stem-vertebrate. However, at this point we cannot reliably establish whether these innovations pre- or postdate the 1R event. This basic vertebrate bodyplan was further elaborated independently in cyclostomes and gnathostomes as a result of their lineage-specific genome duplications, for instance, facilitating the evolution of different adaptive immune systems (immunoglobulin-based in jawed vertebrates, variable lymphocyte receptor-based in cyclostomes35), or the appearance of key morphological innovations, such as the jaw and paired appendages in gnathostomes. Interestingly, these independent WGD events shaped their ancestral genomes in similar ways by permitting an increase in regulatory complexity, especially of genes with roles in development. Duplicates of developmental genes are indeed more likely to be retained in both lineages, highlighting the crucial role of development in evolution of novel complex traits.
The contrasting morphological phenotypic consequences of 2R versus the other WGD events might suggest that there is no direct causal relationship or that there should be no general expectation of macroevolutionary consequences from WGD events despite their clear impact in increasing the regulatory potential of the genome. Another possibility is that the 2R event is different in nature from the 1R and CR events. Indeed, a number of recent studies together with our own analyses (Extended Data Fig. 5b) have suggested that while 1R was probably an autopolyploidy event, 2R was an allopolyploidy11,12,57. This is significant since it impacts our interpretation of the absolute timing of 2R, with the age estimate reflecting the divergence of the two lineages that later hybridized, not the allopolyploidy event itself55. Thus, the event occurred later than 498.4–485.2 Ma (late Cambrian), potentially coinciding with or even postdating the acquisition of gnathostome novelties that accrued among ostracoderms4,58 before the divergence of crown-gnathostomes (which definitively postdated 2R) at 450.1–445.1 Ma (Late Ordovician). By the same token, the macroevolutionary consequences of allopolyploidy are expected to be more immediate than those of autopolyploidy, resulting in chromosomal rearrangements, changes in chromatin structure, DNA methylation, gene expression and the activation of transposable elements59,60,61, extensive and immediate changes that promote species, and ecological diversification62,63 as well as evolutionary novelty64,65,66,67. This may go some way to explain why the evolutionary consequences of the 2R WGD are so much greater, leading to the profound diversification of gnathostome bodyplans that have dominated vertebrate communities since the early Palaeozoic.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment.
Animal sampling and experimentation
Adult inshore hagfish animals were captured off the coast of Shimane, Japan, as previously described68. Hagfish embryos (staged according to ref. 48) used for ATAC-seq were obtained as previously described19,68. The sampling and experiments were conducted according to institutional and national guidelines for animal ethics, approved by the RIKEN Animal Experiments Committee (approvals H14-25-23 and H14-25-25).
Genome sequencing and assembly
We sequenced a mix of short-insert paired-end and long-insert mate pair libraries prepared from DNA extracted from the testis of a single, sexually mature male individual of the inshore hagfish, E. burgeri, resulting in ~240X of Illumina clean data (Supplementary Tables 2 and 3). Hagfish species have large genome sizes, ranging between ~2.2 and 4.5 Gb23. We estimated the genome of E. burgeri at 3.12 Gb on the basis of k-mer frequency distribution (Extended Data Fig. 1a and Supplementary Table 4), in line with other hagfish species. We assembled the genome of E. burgeri following gradual steps using different strategies. First, we obtained a primary assembly using just the Illumina short-read data (v.2.0). To improve contiguity, this primary assembly was super-scaffolded using Chicago in vitro proximity ligation at Dovetail Genomics22, significantly increasing the scaffold N50 (scaffolds equal to or longer than this value contain 50% of the assembly) from 0.44 to 2.69 Mb. This assembly was polished with all short-insert sequencing data using Pilon69 v.1.22, and the resulting version (3.2 in our pipeline) was made publicly available in both NCBI (GenBank accession no. GCA_900186335.2) and Ensembl70 (release 93; https://www.ensembl.org/Eptatretus_burgeri/). We further sequenced over 2200X of raw Hi-C short-read data from a second adult male individual and obtained ~350X valid Hi-C contact data to improve scaffolding. Hi-C contacts were also used to correct 280 likely misjoined scaffolds (Supplementary Table 7). After a process of parameter optimization, we used LACHESIS71 to assemble 1,573 scaffolds into 19 Hi-C contact clusters.
RNA sequencing
Adult tissues were dissected from two adult male individuals of E. burgeri (brain, gills, liver, intestine, heart, skeletal muscle, kidney and testis from animal #20150825; blood from animal #20150917). Total RNA was extracted using an RNeasy Plus Universal mini kit (QIAGEN) for the brain, heart, skeletal muscle, kidney and testis samples, and with ISOGEN (Nippon Gene), a guanidinium thiocyanate-phenol-chloroform-based extraction protocol, for the intestine, liver and gill samples. In all cases, DNA was removed including a DNaseI step. RNA-seq libraries were prepared with the TruSeq Stranded RNA Lib Prep kit (Illumina) and quantified by qPCR using the KAPA Library Quantification kit for Illumina Libraries (KapaBiosystems) for all samples. Library profiles were assessed with an Agilent 2100 Bioanalyzer. All libraries were sequenced at RIKEN BDR in an Illumina HiSeq 1500 platform, obtaining a total of ~650 M 127-bp paired-end strand-specific reads, with an average of ~54.5 M reads per tissue.
Genome annotation
Annotation of the hagfish genome assembly v.3.2 was created via the Ensembl gene annotation system24, assisted by RNA-seq data from 9 adult tissues (this study) and by developmental RNA-seq data from three embryos (Dean stages 35, 40 and 45) generated in a previous study19. Coordinates of annotated features were later converted to the final Hi-C assembly, v.4.0. Detailed methodology and annotation results can be found in Ensembl (http://www.ensembl.org/info/genome/genebuild/2018_06_eptatretus_burgeri_genebuild.pdf) and in Supplementary Information (section 1.2). In addition to the Ensembl pipeline, miRNA genes were further annotated using MirMachine72 (v.0.1.2) and MirMiner73 (v.1.0). Before performing phylogenetic analyses corresponding to Fig. 4a, 1,957 gene models were manually corrected, with the numbers ranging between 120 (amphioxus) and 704 (sea lamprey).
GC content, codon usage and amino acid composition
Overall GC-content percentage was analysed for whole genomes of the inshore hagfish and 9 other chordate genomes (human, Homo sapiens; chicken, Gallus gallus; tropical clawed frog, Xenopus tropicalis; zebrafish, Danio rerio; spotted gar, Lepisosteus oculatus; elephant shark, Callorhinchus milii; sea lamprey, Petromyzon marinus; sea squirt, Ciona robusta; and the Floridian lancelet, Branchiostoma floridae) (Supplementary Table 12). GC-content distribution (Extended Data Fig. 1d) was calculated from non-overlapping sliding 10-kb windows. To calculate codon type frequency, we categorized each codon into GC-0/1/2/3 on the basis of the number of G or C bases in a codon. The summed frequency of usage for each category is the sum of the normalized frequency of codon usage for all codons included in each category. To plot the distribution of GC content per codon position, the GC percentage of each codon position for each protein-coding gene (with only the longest coding sequence per gene) was calculated, as well as the GC content for each whole coding sequence (equivalent to the GC content of all three codon positions). RSCU calculates the relative synonymous codon usage on degenerative sites of third codon positions, which is independent from the amino acid usage. Therefore, RSCU was used as a robust measurement of GC bias in codons. Correspondence analysis of RSCU values was performed with codonW according to ref. 74.
Completeness evaluation of genome and annotation
We used BUSCO75 v.5.2.2 to assess the completeness of genomes at both assembly and annotation levels of hagfish (E. burgeri), three lamprey species (Far Eastern brook lamprey, Lethenteron reissneri76; sea lamprey, P. marinus45; and Arctic lamprey, L. camtschaticum11) and two jawed vertebrates (elephant shark, C. milii11; and chicken, G. gallus v.7.0, downloaded from Ensembl 109). The programme was run in both ‘genome’ and ‘protein’ modes, with gene predictor ‘metaeuk’ against the core metazoan database embedded in BUSCO (metazoa_odb10 dataset, built on 17 February 2021, with 954 BUSCOs).
Species tree inference
Orthogroups of protein-coding genes previously used in the analysis of the spotted gar genome40 were extended using HaMStR77 (v.13.2.6). The spotted gar genes from ref. 40 were used as bait sequences in HaMStR, which sequentially added the best matching protein sequence for each species, provided the bait sequence was in turn the best match in the spotted gar proteome (reciprocity was fulfilled). HaMStR uses hidden Markov model profiles to assign similarity scores. Of the 242 alignments used in the spotted gar study, 190 remained single copy in all the taxa used here. These were used to reconstruct the topology of vertebrates. The 190 protein families were individually aligned using MAFFT78 (v.7.402) with default settings, concatenated to form an alignment of 310,527 sites and trimmed with automatic method selection in trimAl79 v.1.2 (-automated1). This concatenated alignment containing 84,017 sites is available in Supplementary File 13. This was used to infer a phylogeny (also provided in Supplementary File 13). We used PhyloBayes80,81 v.4.1 with the CAT82 GTR83 model with 4 discrete gamma categories for site rates84. The analysis can be repeated in PhyloBayes with: phylobayes – pb -d alignment -cat -gtr -dgam 4. Convergence was analysed visually in Tracer85 (v1.7.1) and using bpcomp and tracecomp in the PhyloBayes suite. Six chains were run for between 12,991 and 13,836 cycles. After a burn-in of 1,500 cycles, bpcomp revealed that all bipartitions were present in exactly the same frequencies (maxdiff and meandiff = 0). Tracecomp revealed effective sample sizes of parameters ranging from 522 to 11,491, with relative differences of 0.018 to 0.218. We deemed that, at least for topology construction, these chains had converged sufficiently, therefore, recovered topologies reflected the true posterior distribution.
Dating species divergences
For molecular clock analysis, we expanded the dataset to include several non-vertebrate outgroups because many of the calibrations have similar maximum bounds, meaning the effective time prior would be older than intended if we did not include the outgroups. HaMStR was then used to extend the orthogroups to include the new taxa. Of the original 190 orthogroups, 172 were retained in single copy in all taxa; they were aligned and trimmed as before. This alignment (provided in Supplementary File 14) was used as input to MCMCtree84 (v.4.9j) using approximate likelihood estimation86. The analysis was run on each gene under the simplest possible model. The temporary control files were then used as input to CODEML (v.4.9j) for each gene with the following modifications. The substitution model was changed to the one that was preferred by ProtTest87 (v.3.4.2) from a subset of LG88, WAG89, JTT90, Dayhoff91 and BLOSUM62 (ref. 92). Fix_alpha was set to 0, alpha was set to 0.5 and the number of gamma categories was set to 5. The Hessian matrices generated were concatenated to form the .BV file, which was used for the approximate likelihood estimation in the full analysis. The time prior was constructed by applying a uniform prior distribution with a hard minimum bound and a soft maximum bound (with 2.5% probability greater than the maximum) to nodes. We used the autocorrelated rates clock model with a gamma prior distribution with shape = 2 and scale = 4.53. This was constructed by dividing a typical distance between two tips whose most recent common ancestor was at the root of the tree under LG + F + G4 (inferred with IQ-TREE93 v.1.6.3) by the expected time for the tree based on the root prior. This was multiplied by the shape parameter of 2 (leading to a fairly flat gamma distribution, corresponding to a relatively uninformative prior). The variance prior (sigma2) had shape = 1 and scale = 1, meaning that variation in rates is not highly penalized in the posterior distribution. Rates across sites were modelled by a gamma distribution with shape = 1 and scale = 1 with 5 discrete categories. After a burn-in period of 10,000 generations, parameter values were saved every 20th generation until 20,000 cycles were saved (400,000 generations in total). Convergence was investigated in Tracer85, revealing convergence had been reached in the six chains run (the lowest effective sample size was 194 and posterior distributions in all 6 chains looked almost identical). The alignments, control files and tree are available in Supplementary File 14.
Estimation of gene duplication rates
Orthogroups were predicted using OrthoFinder94,95 v.2.3.5; output from this analysis is available as Supplementary File 15. OrthoFinder includes a gene duplication prediction step as part of its pipeline. Gene duplication events presented here had >50% support. The species tree was fixed to the topology inferred in this study.
Rooting the vertebrate phylogeny
Orthogroups were predicted using OrthoFinder94 v.2.3.5 for only the vertebrate taxa (hagfish, lampreys and gnathostomes). For each gene family, sequences were aligned using MAFFT78 (v.7.402) with default settings, then trimmed using trimAl79 with heuristic choice of trimming parameters. IQ-TREE93 was then used to generate 1,000 bootstrapped trees in a maximum likelihood framework, with the model selected using ModelFinder96 (as part of IQ-TREE v.1.6.3). These bootstrapped trees were used as the input to ALEobserve (v.1.0) to create ALE objects. Two species trees were used as hypotheses; one with hagfish as sister to all other vertebrates and one with monophyletic cyclostomes. ALEml_undated29 (v.1.0) was used with each of these species tree hypotheses with default settings, except for tau (the transfer rate) which was set to 0, meaning that transfers could not be inferred. This estimates the pattern of gene duplication and loss for each gene family under the different species tree hypotheses, as well as a likelihood under the species tree. An approximately unbiased test30 was then performed on the likelihoods of each gene family under the two competing hypotheses using the programme CONSEL97 (v.0.2.0).
Ancestral gene family complements
A total of 45 animal genomes (Supplementary Table 1 and Extended Data Fig. 3) were compared using a pipeline described previously31,32,33. Briefly, the proteomes were compared using a reciprocal blastp of all-vs-all sequences with DIAMOND98 (v.0.9.30.131; e-value threshold of 1 × 10−5). Markov cluster algorithm99 (v.1:14-137+ds-4) was used to infer homology groups (HGs) from the BLAST output with default inflation parameter (I = 2). GOs were assigned to the different HGs by analysing the human protein sequences in each HG with PANTHER GO100 (v.15.0).
Orthology relationships of Hox gene clusters
Hagfish Hox sequences were obtained from a previous study19 and used as queries in TBLASTN (v.2.10.1+) to find the location in the Hi-C assembly and Ensembl annotation. Information about Hox syntenic genes in lamprey, human and elephant shark, with the European amphioxus as outgroup, were obtained from previous studies17,18,19,101, downloaded from Ensembl or NCBI GenBank and used as queries to find their presence in the hagfish Hi-C assembly and Ensembl annotation in TBLASTN. Location of Hox and their syntenic genes, as well as their Ensembl Gene IDs is provided in Supplementary Table 29. For phylogenetic analysis of Hnrnpa, Cbx, Gbx and Agap, amino acid sequences were aligned using MUSCLE102 as implemented in MEGAX103 v.10.2.4. The alignment was trimmed by trimAl79 v.1.2rev59 using the ‘-automated1’ option and then formatted into a nexus file using readAl (bundled with the trimAl package). The Bayesian inference tree was constructed using MrBayes104 v.3.2.6, under the assumption of an LG + I + G evolutionary model, with two independent runs and four chains. The tree was considered to have reached convergence when the standard deviation stabilized under a value of <0.01. A burn-in of 25% of the trees was performed to generate consensus trees. Multisequence alignments with MrBayes parameters and number of generations for each tree are provided in Supplementary Files 16–19.
Dating genome duplications in vertebrates
OrthoFinder-inferred gene families were selected that showed a clear signal of both the 1R and 2R duplication events and were broadly congruent with current phylogenetic hypotheses. This resulted in 35 gene families in which each gnathostome was represented up to four times and each cyclostome twice. Gene families containing a signal of both 1R, 2R and the cyclostome duplication event (CR) were rare, hence, to date the CR event, an additional dataset was assembled consisting of 27 gene families in which each cyclostome species was represented by at least two gene copies.
For each analysis, taxon sampling towards the root of the tree was improved by including additional outgroup taxa Nematostella vectensis (Cnidaria), Trichoplax adhaerens (Placozoa), Mnemiopsis leidyi (Ctenophora) and Hofstenia miamia (Xenacoelomorpha); this served to remove the nodes of interest from the root of the tree and include additional relative and absolute calibration information for more universal clades. Individual gene families were aligned using MUSCLE102 (v.5) and trimmed using the ‘-automated1’ option in trimAl79. The best-fitting model for each gene family was determined using IQ-TREE93 (v.2.1.3) and all gene families were concatenated into a single alignment.
The node age time priors were based on the posterior estimates from the associated species divergence times analysis (see ‘Dating species divergences’ above), using the span of the 95% highest density credibility intervals of node ages from that analysis to inform uniform time priors on the same species nodes in gene tree analysis, with a 1% probability tail that the maximum age could be exceeded. Calibrations within lineages that have undergone WGD were repeated across the duplicated clades with identical probability distributions. Molecular clock analyses were performed using the normal approximation method in MCMCtree105 (v.4.9j), with each gene treated as a separate partition. Four independent Markov chain Monte Carlo (MCMC) chains were run for 2 million generations each, with the first 20% discarded as burn-in. Convergence was determined using Tracer85 and by comparing congruence among all four runs. The alignments, MCMCtree control files and calibrations used are available in Supplementary File 20; the dates of species divergence are presented in Fig. 2; and dating of WGD events is shown in Fig. 2 and Supplementary Fig. 34.
Reconstruction of vertebrate ancestral chromosomes
On the basis of reciprocal best BLASTP v.2.6.0+ (e-value threshold of 1 × 10−6) search and a chi-squared test (multiple test correction with false discovery rate, q value threshold of 0.05), we identified homologous chromosomes within either the chicken or spotted gar genome that possessed significantly more between-chromosome homologous genes. Homologous chromosomes between either the chicken or spotted gar genome and sea cucumber chromosomes were also inferred, except that the best BLASTP search was unidirectional wherein the sea cucumber genes were the reference. From inferred within-species and between species homologies, all chicken and spotted gar chromosomes were grouped into 17 groups representing the 17 predicted ACs that contribute to extant gnathostome karyotypes. The gene content of these ACs was reconstructed with Belcher’s lancelet (B. belcheri) genes43. A Belcher’s lancelet gene was distributed to one vertebrate ancestral chromosome if either (1) the scaffold this gene is located on is a homologous scaffold to the specific sea cucumber linkage group, and this gene is homologous to the corresponding chicken and spotted gar genes; or (2) this gene is homologous to at least five different chicken and spotted gar genes. As a result, 5,065 lancelet genes were anchored to the 17 inferred ACs and used as their gene content, ranging from 115 to 534 genes in ACs 1–16, and only 20 genes in AC17 (Supplementary Table 34).
Phylogenetic support around 1R/2R
A homologous gene set is a group of genes that share the same best BLASTP hit AC gene. Multiple sequence alignments were obtained with PRANK106 v.150803. ModelFinder96 (embedded in IQ-TREE93 v.1.6.12) with BIC criteria and ‘-mtree’ parameter was used to find the best-fitting model. RAxML-ng v.0.9.0 and IQ-TREE were repeatedly run for 10 times with different seed numbers. For the 20 obtained maximum likelihood trees, we used RAxML-ng (v.0.9.0) to re-evaluate their likelihoods and chose the best tree as the final tree for each homologous gene set (Supplementary Files 5–8).
Definition and calculation of overlapping ratio
For a reference chromosome and all genes on it, the existence or absence of a homologue on a query chromosome is denoted as binary mode 1 or 0. We defined it as the gene retention profile. Mathematically, it is a vector with values of either 1 or 0 and with fixed length that corresponds to the number of genes on the query chromosome. One notable property of the gene retention profile is that the gene order within the query chromosome does not alter the gene retention profile itself. The OR was calculated between two gene retention profiles that correspond to one same reference chromosome. It is equal to the number of shared homologues divided by the smaller one of two total numbers of homologues and has a value range between 0 and 1. Notably, the OR is insensitive to the size difference between two query chromosomes.
Hierarchical clustering based on the gene retention profile
For multiple chromosomes homologous to one same vertebrate ancestral chromosome, we inferred their gene retention profiles and calculated all pairwise ORs. We used 1 − OR as a measure of pairwise distance and performed hierarchical clustering with the ‘Ward.D’ method provided in the R platform.
GO enrichment analysis of ohnologues
We mapped hagfish and chicken ohnologues to human genes and performed GO enrichment analysis with human orthologues. Functional enrichment was examined with the Metascape107 online tool. We used the 959 human orthologues of hagfish ohnologues and randomly sampled 2,999 genes (as Metascape has a limit of 3,000) from a total of 3,595 chicken orthologues. GO (biological process) enrichment analysis was performed against all genes of the two species. Genes were annotated as either developmental ohnologues, non-developmental ohnologuess or non-ohnologous genes according to their GO terms annotated by PANTHER108 v.17.0.
Chromatin accessibility profiling
ATAC-seq experiments on two hagfish embryos at stages Dean 45 (collected in 2018) and 53 (collected in 2017) (Supplementary Fig. 31) were performed following previous descriptions101,109 with slight variations (details are provided in Supplementary Information, section 5). Embryos were divided and processed into two halves to gain positional information to be used in a future project. Approximately 50,000 nuclei per replicate (~200,000 nuclei per embryo) were processed for tagmentation using Tn5 from the Illumina Nextera DNA Library Prep kit. Libraries were multiplexed and sequenced at the Beijing Genomic Institute in 4 lanes (2 per embryo) in an Illumina HiSeq 4000 platform.
To identify ACRs as putative gene cis-regulatory regions, we collected ATAC-seq data of hagfish and other chordate embryos (amphioxus, zebrafish, medaka, chicken and mouse; GSE106428 (ref. 47) and DRA006971 (ref. 110)) with two replicates each (Supplementary Table 51). For each data, ATAC-seq paired-end reads were aligned to the reference genome using Bowtie2 (ref. 111) (v.2.4.2). After extracting nucleosome-free read pairs (the insert shorter than 120 bp), we performed peak-calling by using MACS2 (ref. 112) (v.2.2.7.1). Finally, on the basis of the replicate information, reproducible peaks were identified as ACRs using the IDR framework113.
Fate of ohnologues after WGD
After quantile normalization, transcripts per million >5 was used as a threshold to consider a gene to have either an ‘expressed’ or ‘not expressed’ state (Supplementary Fig. 31). Only ohnologue pairs in which both genes are expressed in at least one tissue were analysed. Fates of ohnologues were classified according to their expressional patterns in the tissues assayed in this study for the hagfish, or from a previous study in the case of chicken114. Fates were defined following a different strategy from that in ref. 47 due to the lack of information from homologous tissues of the amphioxus (outgroup). After WGD, ohnologues can follow one of the following fates: (1) potential redundancy, if the two ohnologues are expressed in the same set of tissues; (2) potential subfunctionalization, if both ohnologues are each expressed in a tissue not shared with the other. In other words, each of them has tissue-specific expression domains; (3) potential specialization, if one ohnologue has a reduced set of expression domains contained in a larger set of tissues in which the other ohnologue is expressed. Gene families within ‘specialization’ can be further defined as having either ‘potential strong specialization’ when the ohnologue with the narrower expression pattern is transcribed in <40% of the domains than the ohnologue with the broader expression pattern; or ‘potential mild specialization’ when the ohnologue with the narrower expression pattern is transcribed in ≥40% of domains than the ohnologue with the broader expression pattern.
Phenotypic disparity analyses
A character matrix of 578 characters and 278 taxa was assembled as follows. Characters were collected from direct observations and multiple literature sources115,116,117,118,119,120,121,122,123,124,125,126. Previous literature sources were modified to ensure that duplicated character states were removed and that overlapping characters from different sources were combined into single characters or subdivided into multiple characters to encompass all variation across vertebrates. We ensured that all characters were coded for as many taxa as possible. Missing data are coded as ‘?’; inapplicable characters are coded as ‘-’. Character state observations were coded using primary observations and through the literature. Characters were coded using hierarchical contingencies127,128,129. The character matrix and descriptions are available in Supplementary File 21 (Vertebrate_disparity_matrix.nex).
The phenotype character matrix was transformed before disparity analyses such that characters coded ‘not applicable’ were scored as ‘0’ and each subsequent character state was increased by 1. Ancestral character states were estimated along a tree representative of current phylogenetic hypotheses using stochastic character mapping130, with 1,000 simulations per character; the tree is available in Supplementary File 21 (Disparity.tre). Distances between taxa and reconstructed internal nodes were estimated using Gower’s dissimilarity metric131, and these distances were ordinated using NMDS, a method that seeks to reduce dimensionality while preserving distances between taxa. A pre-ordination phylomorphospace was plotted using the inferred ancestral states, NMDS scores and the representative phylogeny. Convex hulls were fitted around taxonomic lineages and groups that have undergone successive rounds of WGD. All stem gnathostomes were adjudged to have undergone the 2R WGD because they postdate the timing of 2R inferred from the gene tree-based molecular clock analysis (see ‘Dating genome duplications in vertebrates’ above). Disparity metrics were estimated using dispRity (v.1.7.0) in R (v.2.6-4) with 1,000 bootstrap replicates132.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Eptatretus burgeri (inshore hagfish) v.4.0 genome is available in NCBI GenBank under accession number GCA_900186335.3. Raw genome sequencing data together with adult RNA-seq data have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB21290. ATAC-seq data have been deposited in Gene Expression Omnibus (GEO) under accession number GSE247552. Supplementary files are available at FigShare (https://figshare.com/projects/Hagfish_Genome_Project/163186). Gene annotation used in this study is available at https://www.ensembl.org/Eptatretus_burgeri. A mirror of the UCSC Genome Browser containing hagfish assembly and annotations is available at http://ucsc.crg.eu/.
References
Van de Peer, Y., Maere, S. & Meyer, A. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 10, 725–732 (2009).
Dehal, P. & Boore, J. L. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 3, e314 (2005).
Ohno, S. Evolution by Gene Duplication (Springer, 1970).
Donoghue, P. & Purnell, M. Genome duplication, extinction and vertebrate evolution. Trends Ecol. Evol. 20, 312–319 (2005).
Holland, L. Z. & Ocampo Daza, D. A new look at an old question: when did the second whole genome duplication occur in vertebrate evolution? Genome Biol. 19, 2–5 (2018).
Shimeld, S. M. & Donoghue, P. C. J. Evolutionary crossroads in developmental biology: cyclostomes (lamprey and hagfish). Development 139, 2091–2099 (2012).
Kuraku, S., Meyer, A. & Kuratani, S. Timing of genome duplications relative to the origin of the vertebrates: did cyclostomes diverge before or after? Mol. Biol. Evol. 26, 47–59 (2009).
Sacerdot, C., Louis, A., Bon, C., Berthelot, C. & Roest Crollius, H. Chromosome evolution at the origin of the ancestral vertebrate genome. Genome Biol. 19, 166 (2018).
Escriva, H., Manzon, L., Youson, J. & Laudet, V. Analysis of lamprey and hagfish genes reveals a complex history of gene duplications during early vertebrate evolution. Mol. Biol. Evol. 19, 1440–1450 (2002).
Holland, P. W. H., Garcia-Fernandez, J., Williams, N. A. & Sidow, A. Gene duplications and the origins of vertebrate development. Development 120, 125–133 (1994).
Nakatani, Y. et al. Reconstruction of proto-vertebrate, proto-cyclostome and proto-gnathostome genomes provides new insights into early vertebrate evolution. Nat. Commun. 12, 4489 (2021).
Simakov, O. et al. Deeply conserved synteny resolves early events in vertebrate evolution. Nat. Ecol. Evol. 4, 820–830 (2020).
Putnam, N. H. et al. The amphioxus genome and the evolution of the chordate karyotype. Nature 453, 1064–1071 (2008).
Nakatani, Y., Takeda, H., Kohara, Y. & Morishita, S. Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res. 17, 1254–1265 (2007).
Lamb, T. D. Analysis of paralogons, origin of the vertebrate karyotype, and ancient chromosomes retained in extant species. Genome Biol. Evol. 13, evab044 (2021).
Smith, J. J. & Keinath, M. C. The sea lamprey meiotic map improves resolution of ancient vertebrate genome duplications. Genome Res. 25, 1081–1090 (2015).
Smith, J. J. et al. The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat. Genet. 50, 270–277 (2018).
Mehta, T. K. et al. Evidence for at least six Hox clusters in the Japanese lamprey (Lethenteron japonicum). Proc. Natl Acad. Sci. USA 110, 16044–16049 (2013).
Pascual-Anaya, J. et al. Hagfish and lamprey Hox genes reveal conservation of temporal colinearity in vertebrates. Nat. Ecol. Evol. 2, 859–866 (2018).
Smith, J. J., Antonacci, F., Eichler, E. E. & Amemiya, C. T. Programmed loss of millions of base pairs from a vertebrate genome. Proc. Natl Acad. Sci. USA. 106, 11212–11217 (2009).
Kohno, S., Kubota, S. & Nakai, Y. in The Biology of Hagfishes 81–100 (Springer, 1998).
Putnam, N. H. et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350 (2016).
Kuraku, S. Insights into cyclostome phylogenomics: pre-2R or post-2R. Zool. Sci. 25, 960–968 (2008).
Aken, B. L. et al. The Ensembl gene annotation system. Database 2016, baw093 (2016).
Heimberg, A. M., Cowper-Sal·lari, R., Sémon, M., Donoghue, P. C. J. & Peterson, K. J. microRNAs reveal the interrelationships of hagfish, lampreys, and gnathostomes and the nature of the ancestral vertebrate. Proc. Natl Acad. Sci. USA 107, 19379–19383 (2010).
Fromm, B. et al. MirGeneDB 2.1: toward a complete sampling of all major animal phyla. Nucleic Acids Res. 50, D204–D210 (2022).
Kuraku, S. & Kuratani, S. Time scale for cyclostome evolution inferred with a phylogenetic diagnosis of hagfish and lamprey cDNA sequences. Zool. Sci. 23, 1053–1064 (2006).
Kuratani, S. in Current Topics in Developmental Biology Vol. 141 (ed. Gilbert, S. F.) 207–239 (Academic Press, 2021).
Szöllősi, G. J., Rosikiewicz, W., Boussau, B., Tannier, E. & Daubin, V. Efficient exploration of the space of reconciled gene trees. Syst. Biol. 62, 901–912 (2013).
Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 51, 492–508 (2002).
Paps, J. & Holland, P. W. H. Reconstruction of the ancestral metazoan genome reveals an increase in genomic novelty. Nat. Commun. 9, 1730 (2018).
Bowles, A. M. C., Bechtold, U. & Paps, J. The origin of land plants is rooted in two bursts of genomic novelty. Curr. Biol. 30, 530–536.e2 (2020).
Guijarro-Clarke, C., Holland, P. W. H. & Paps, J. Widespread patterns of gene loss in the evolution of the animal kingdom. Nat. Ecol. Evol. 4, 519–523 (2020).
Tan, M. et al. The whale shark genome reveals patterns of vertebrate gene family evolution. eLife 10, e65394 (2021).
Boehm, T. et al. Evolution of alternative adaptive immune systems in vertebrates. Annu. Rev. Immunol. 36, 19–42 (2018).
Meyer, A. & Van de Peer, Y. From 2R to 3R: evidence for a fish-specific genome duplication (FSGD). BioEssays 27, 937–945 (2005).
Nakatani, Y. & McLysaght, A. Macrosynteny analysis shows the absence of ancient whole-genome duplication in lepidopteran insects. Proc. Natl Acad. Sci. USA 116, 1816–1818 (2019).
Simakov, O. et al. Deeply conserved synteny and the evolution of metazoan chromosomes. Sci. Adv. 8, eabi5884 (2022).
Hillier, L. W. et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695–716 (2004).
Braasch, I. et al. The spotted gar genome illuminates vertebrate evolution and facilitates human–teleost comparisons. Nat. Genet. 48, 427–437 (2016).
Venkatesh, B. et al. Elephant shark genome provides unique insights into gnathostome evolution. Nature 505, 174–179 (2014).
Zhang, X. et al. The sea cucumber genome provides insights into morphological evolution and visceral regeneration. PLOS Biol. 15, e2003790 (2017).
Huang, S. et al. Decelerated genome evolution in modern vertebrates revealed by analysis of multiple lancelet genomes. Nat. Commun. 5, 5896 (2014).
Parey, E. et al. An atlas of fish genome evolution reveals delayed rediploidization following the teleost whole-genome duplication. Genome Res. 32, 1685–1697 (2022).
Timoshevskaya, N. et al. An improved germline genome assembly for the sea lamprey Petromyzon marinus illuminates the evolution of germline-specific chromosomes. Cell Rep. 42, 112263 (2023).
Redmond, A. K., Casey, D., Gundappa, M. K., Macqueen, D. J. & McLysaght, A. Independent rediploidization masks shared whole genome duplication in the sturgeon-paddlefish ancestor. Nat. Commun. 14, 2879 (2023).
Marlétaz, F. et al. Amphioxus functional genomics and the origins of vertebrate gene regulation. Nature 564, 64–70 (2018).
Oisi, Y., Ota, K. G., Kuraku, S., Fujimoto, S. & Kuratani, S. Craniofacial development of hagfishes and the evolution of vertebrates. Nature 493, 175–180 (2013).
Sandve, S. R., Rohlfs, R. V. & Hvidsten, T. R. Subfunctionalization versus neofunctionalization after whole-genome duplication. Nat. Genet. 50, 908–909 (2018).
Álvarez-Carretero, S. et al. A species-level timeline of mammal evolution integrating phylogenomic data. Nature 602, 263–267 (2022).
Gundappa, M. K. et al. Genome-wide reconstruction of rediploidization following autopolyploidization across one hundred million years of salmonid evolution. Mol. Biol. Evol. 39, msab310 (2022).
Lien, S. et al. The Atlantic salmon genome provides insights into rediploidization. Nature 533, 200–205 (2016).
Martin, K. J. & Holland, P. W. H. Enigmatic orthology relationships between Hox clusters of the African butterfly fish and other teleosts following ancient whole-genome duplication. Mol. Biol. Evol. 31, 2592–2611 (2014).
Robertson, F. M. et al. Lineage-specific rediploidization is a mechanism to explain time-lags between genome duplication and evolutionary diversification. Genome Biol. 18, 111 (2017).
Doyle, J. J. & Egan, A. N. Dating the origins of polyploidy events. New Phytol. 186, 73–85 (2010).
Shimeld, S. M. & Holland, P. W. H. Vertebrate innovations. Proc. Natl Acad. Sci. USA 97, 4449–4452 (2000).
Peterson, K. J. et al. MicroRNAs as indicators into the causes and consequences of whole-genome duplication events. Mol. Biol. Evol. 39, msab344 (2022).
Donoghue, P. C. J. & Keating, J. N. Early vertebrate evolution. Palaeontology 57, 879–893 (2014).
Kon, T. et al. The genetic basis of morphological diversity in domesticated goldfish. Curr. Biol. 30, 2260–2274.e6 (2020).
Qiu, T., Liu, Z. & Liu, B. The effects of hybridization and genome doubling in plant evolution via allopolyploidy. Mol. Biol. Rep. 47, 5549–5558 (2020).
Soltis, P. S. & Soltis, D. E. Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 30, 159–165 (2016).
Barker, M. S., Arrigo, N., Baniaga, A. E., Li, Z. & Levin, D. A. On the relative abundance of autopolyploids and allopolyploids. New Phytol. 210, 391–398 (2016).
Estep, M. C. et al. Allopolyploidy, diversification, and the Miocene grassland expansion. Proc. Natl Acad. Sci. USA 111, 15149–15154 (2014).
Alix, K., Gérard, P. R., Schwarzacher, T. & Heslop-Harrison, J. S. (Pat).Polyploidy and interspecific hybridization: partners for adaptation, speciation and evolution in plants. Ann. Bot. 120, 183–194 (2017).
Steensels, J., Gallone, B. & Verstrepen, K. J. Interspecific hybridization as a driver of fungal evolution and adaptation. Nat. Rev. Microbiol. 19, 485–500 (2021).
Taylor, S. A. & Larson, E. L. Insights from genomes into the evolutionary importance and prevalence of hybridization in nature. Nat. Ecol. Evol. 3, 170–177 (2019).
Wu, Y. et al. Genomic mosaicism due to homoeologous exchange generates extensive phenotypic diversity in nascent allopolyploids. Natl Sci. Rev. 8, nwaa277 (2021).
Oisi, Y., Kakitani, O., Kuratani, S. & Ota, K. G. in In Situ Hybridization Methods Vol. 99 (ed. Hauptmann, G.) 249–262 (Springer, 2015).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Cunningham, F. et al. Ensembl 2019. Nucleic Acids Res. 47, D745–D751 (2019).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Umu, S. U. et al. Accurate microRNA annotation of animal genomes using trained covariance models of curated microRNA complements in MirMachine. Cell Genomics 3, 100348 (2023).
Wheeler, B. M. et al. The deep evolution of metazoan microRNAs. Evol. Dev. 11, 50–68 (2009).
Smith, J. J. et al. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat. Genet. 45, 415–421 (2013).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Zhu, T. et al. Chromosome‐level genome assembly of Lethenteron reissneri provides insights into lamprey evolution. Mol. Ecol. Resour. 21, 448–463 (2021).
Ebersberger, I., Strauss, S. & von Haeseler, A. HaMStR: profile hidden Markov model based search for orthologs in ESTs. BMC Evol. Biol. 9, 157 (2009).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Lartillot, N., Brinkmann, H. & Philippe, H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol. Biol. 7, S4 (2007).
Lartillot, N. & Philippe, H. Computing Bayes factors using thermodynamic integration. Syst. Biol. 55, 195–207 (2006).
Lartillot, N. & Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 21, 1095–1109 (2004).
Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 17, 57–86 (1986).
Yang, Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 39, 306–314 (1994).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018).
dos Reis, M. & Yang, Z. Approximate likelihood calculation on a phylogeny for Bayesian estimation of divergence times. Mol. Biol. Evol. 28, 2161–2172 (2011).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 (2011).
Le, S. Q. & Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320 (2008).
Whelan, S. & Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18, 691–699 (2001).
Jones, D. T., Taylor, W. R. & Thornton, J. M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 8, 275–282 (1992).
Dayhoff, M. O., Schwartz, R. M. & Orcutt, B. C. in Atlas of Protein Sequences and Structure (ed. Dayhoff, M. O.) 345–352 (National Biomedical Research Foundation, 1978).
Henikoff, S. & Henikoff, J. G. Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA 89, 10915–10919 (1992).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157 (2015).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Shimodaira, H. Approximately unbiased tests of regions using multistep-multiscale bootstrap resampling. Ann. Stat. 32, 2616–2641 (2004).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Van Dongen, S. Graph clustering via a discrete uncoupling process. SIAM J. Matrix Anal. Appl. 30, 121–141 (2008).
Mi, H., Poudel, S., Muruganujan, A., Casagrande, J. T. & Thomas, P. D. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res. 44, D336–D342 (2016).
Acemel, R. D. et al. A single three-dimensional chromatin compartment in amphioxus indicates a stepwise evolution of vertebrate Hox bimodal regulation. Nat. Genet. 48, 336–341 (2016).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Yang, Z. PAML 4: a program package for phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Löytynoja, A. & Goldman, N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 320, 1632–1635 (2008).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Mi, H. et al. PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 49, D394–D403 (2021).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Uesaka, M., Kuratani, S., Takeda, H. & Irie, N. Recapitulation-like developmental transitions of chromatin accessibility in vertebrates. Zool. Lett. 5, 33 (2019).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Li, Q., Brown, J. B., Huang, H. & Bickel, P. J. Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 5, 1752–1779 (2011).
Brawand, D. et al. The evolution of gene expression levels in mammalian organs. Nature 478, 343–348 (2011).
Brazeau, M. D. The braincase and jaws of a Devonian ‘acanthodian’ and modern gnathostome origins. Nature 457, 305–308 (2009).
Clement, A. M. et al. Neurocranial anatomy of an enigmatic Early Devonian fish sheds light on early osteichthyan evolution. eLife 7, e34349 (2018).
Davis, S. P., Finarelli, J. A. & Coates, M. I. Acanthodes and shark-like conditions in the last common ancestor of modern gnathostomes. Nature 486, 247–250 (2012).
Gabbott, S. E. et al. Pigmented anatomy in Carboniferous cyclostomes and the evolution of the vertebrate eye. Proc. R. Soc. B 283, 20161151 (2016).
Giles, S., Friedman, M. & Brazeau, M. D. Osteichthyan-like cranial conditions in an Early Devonian stem gnathostome. Nature 520, 82–85 (2015).
Keating, J. N. & Donoghue, P. C. J. Histology and affinity of anaspids, and the early evolution of the vertebrate dermal skeleton. Proc. R. Soc. B 283, 20152917 (2016).
Lundgren, M. & Blom, H. Phylogenetic relationships of the cyathaspidids (Heterostraci). GFF 135, 74–84 (2013).
Miyashita, T. et al. Hagfish from the Cretaceous Tethys Sea and a reconciliation of the morphological–molecular conflict in early vertebrate phylogeny. Proc. Natl Acad. Sci. USA 116, 2146–2151 (2019).
Pernègre, V. N. & Elliott, D. K. Phylogeny of the Pteraspidiformes (Heterostraci), Silurian–Devonian jawless vertebrates. Zool. Scr. 37, 391–403 (2008).
Randle, E. & Sansom, R. S. Exploring phylogenetic relationships of Pteraspidiformes heterostracans (stem-gnathostomes) using continuous and discrete characters. J. Syst. Palaeontol. 15, 583–599 (2017).
Sansom, R. S. Phylogeny, classification and character polarity of the Osteostraci (Vertebrata). J. Syst. Palaeontol. 7, 95–115 (2009).
Zhu, M. & Gai, Z. Phylogenetic relationships of galeaspids (Agnatha). Front. Biol. China 2, 151–169 (2007).
Brazeau, M. D. Problematic character coding methods in morphology and their effects. Biol. J. Linn. Soc. 104, 489–498 (2011).
Lee, D. C. & Bryant, H. N. A reconsideration of the coding of inapplicable characters: assumptions and problems. Cladistics 15, 373–378 (1999).
Strong, E. E. & Lipscomb, D. Character coding and inapplicable data. Cladistics 15, 363–371 (1999).
Huelsenbeck, J. P., Nielsen, R., Bollback, J. P. & Schultz, T. Stochastic mapping of morphological characters. Syst. Biol. 52, 131–158 (2003).
Gower, J. C. A general coefficient of similarity and some of its properties. Biometrics 27, 857–874 (1971).
Guillerme, T. & Poisot, T. dispRity: a modular R package for measuring disparity. Methods Ecol. Evol. 9, 1755–1763 (2018).
Pascual-Anaya, J. & Böhmer, C. in Hox Modules in Evolution and Development 121–139 (CRC Press, 2023).
Acknowledgements
This research was supported by grants from the Ministry of Science and Innovation of Spain (PID2021-125078NA-I00) and a KAKENHI Grant-in-aid for Scientific Research C from the Japan Society for the Promotion of Science (19K06798) to J.P.-A.; the National Key R&D Program of China (2019YFA0802600), the Chinese Academy of Sciences (ZDBS-LY-SM005), the National Natural Science Foundation of China (31970565) and the Open Research Program of the Chinese Institute for Brain Research (Beijing) to Y.E.Z.; the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB13000000) to Y.E.Z and W. Wang; the Wellcome Trust (to F.J.M., Grant number 108749/Z/15/Z); the Leverhulme Trust (RF-2022-167 to P.C.J.D); the Biotechnology and Biological Sciences Research Council (BB/T012773/1 to P.C.J.D); the Natural Environment Research Council (NERC) grant (NE/P013678/1 to P.C.J.D. and D.P.), part of the Biosphere Evolution, Transitions and Resilience (BETR) programme, which is co-funded by the Natural Science Foundation of China (NSFC); the John Templeton Foundation (Grant 62220 to P.C.J.D. and D.P.; the opinions expressed in this publication are those of the author(s) and do not necessarily reflect the views of the John Templeton Foundation); Wellcome Trust Seed Award (210101/Z/18/Z to J.P.); the Tromsø Research Foundation (Tromsø Forskningsstiftelse, TFS) to B.F. (20_SG_BF ‘MIRevolution’); a National Science Foundation (NSF) grant (1755418) to M.H.; and National Institutes of Health (NIH) grants (R01AI072435 and R35GM122591) to M.D.C. Computing was jointly supported by the HPC Platform of BIG and that of the Scientific Information Centre of IOZ. We thank O. Kakitani and the members of the Fishery Association in Gotsu City, Shimane Prefecture, Japan, for assistance in collecting hagfish; the technical support staff of the Laboratory for Phyloinformatics in RIKEN Kobe for sequencing data production, and RIKEN aquarium staff; T. de Dios Fernández for animal illustrations. For the purpose of open access, the authors have applied for a CC BY public copyright licence to any author-accepted manuscript version arising from this submission.
Author information
Authors and Affiliations
Contributions
D.Y., Y.R., J.S., Y.L., W. Wan, W. Wang, Y.E.Z. and J.P.-A sequenced, assembled and evaluated the hagfish genome. F.J.M. generated and coordinated the annotation process at Ensembl. M.M., C.C., M.P. and W.A. generated Ensembl Compara data. A.d.M., D.Y., A.J.S.B. and J.W.C. generated orthogroups. R.F., J.F.F., F.S., S.D’A., C.B., C.A., G.S., B.F., K.J.P., S.D., M.H., J.P.R. and M.D.C. contributed to gene family curation. A.J.S.B., J.W.C., D.P. and P.C.J.D. performed phylogenomics and dating analyses. J.P. performed analysis of gene family evolution. D.Y., J.S. and Y.E.Z performed macrosynteny analysis. I.M. and M.I. advised on macrosynteny and ohnologue evolution analyses and discussed the data. J.P.-A, I.S., F.S. and S.K. obtained hagfish embryonic and adult material. J.P.-A performed ATAC-seq and RNA-seq experiments and ensured sequencing project management with BGI and RIKEN. M.U. and J.S. carried out regulatory genome profiling and ohnologue fate evolution analyses. J.N.K., E.M.C., R.P.D., S.G., E.R., R.S.S. and P.C.J.D. obtained fossil and calibration data, and performed morphological disparity analysis. W. Wang, S.K., N.I. and J.P.-A. conceived the project. S.K., N.I., P.C.J.D., W. Wang, Y.E.Z and J.P.-A contributed to project design. J.P.-A. coordinated the project. Y.E.Z., P.C.J.D and J.P.-A. wrote the paper, with inputs from all authors. All authors revised and approved the paper.
Corresponding authors
Ethics declarations
Competing interests
M.D.C. is a cofounder and shareholder of NovAb, Inc., which produces lamprey antibodies for biomedical purposes. J.P.R. is a consultant for NovAb. The other authors declare no competing interests.
Peer review
Peer review information
Nature Ecology & Evolution thanks Aoife McLysaght and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Features of the hagfish genome.
a, 17-mer distribution for inshore hagfish genome size estimation using all raw reads from short insert-size libraries. b, Counts for major classes of genes and transcripts from Ensembl annotation. c, Completeness assessment of the annotation of the inshore hagfish E. burgeri genome (red), three lamprey species (blue) and two jawed vertebrates (green). Numbers of conserved metazoan orthologs (metazoa_odb10 dataset, n = 954) are indicated for each case. F. E., Far Eastern d, GC-content distribution of the hagfish genome and other chordates calculated from 10-kb non-overlapping windows. e, All codon type frequency in given chordate genomes according to GC-content. GC-0/1/2/3 indicates the number of G or C bases in a codon. f, Distribution of GC content at each codon position or at all codon positions (Codon1 + 2 + 3). For each protein coding gene, we only kept the longest coding sequence. For each coding sequence, we calculated the GC content at separate codon positions. We also calculated the GC content for each coding sequence, which is equal to the GC content of all three codon positions. g, Violin plots of size distribution of intron (left), intergenic (middle) and gene body (right) lengths over a logarithmic scale of the hagfish (E. burgeri), two lamprey species (sea lamprey, P. marinus; Arctic lamprey, L. camtschaticum), six gnathostome vertebrates (human, H. sapiens; frog, X. tropicalis; coelacanth, L. chalumnae; chicken, G. gallus; spotted gar, L. oculatus; and the elephant shark, C. milii) and two invertebrate deuterostomes (sea cucumber, A. japonicus; amphioxus, B. belcheri). For each genomic feature and each species, the median (centre line) and IQR (interquartile range) length statistics are indicated with a white rectangle; whiskers extend to the last point no further than 1.5 times the interquartile range from the first and third quartiles Dashed vertical line indicates median size of E. burgeri features; *** P < 2.2 × 10−16, two-sided Wilcoxon rank sum test. Animal illustrations kindly provided by Tamara de Dios Fernández and reproduced with permission from REF. 133.
Extended Data Fig. 2 Phylogenomic analysis of chordates and gene duplication rates across metazoan evolution.
a, Bayesian Inference tree of 31 chordate species was built using a protein alignment (see Methods). All nodes were recovered with a posterior probability of 1. Cyclostome monophyly was unequivocally supported. Scale bar indicates 0.1 substitutions per site. b, c, The exact number of gene duplication events inferred using OrthoFinder2 with greater than 50% support (b) and the number of events per million years per branch (c) are shown. In each case, the colour of the branch represents the value according to the key on the upper-left of each pane. Hagfish is highlighted in red font in all panels.
Extended Data Fig. 3 Reconstruction of ancestral gene content.
a, Cladogram showing the phylogenetic relationships of 45 species representatives of all major eumetazoan taxa with species of gnathostomes and cyclostomes highlighted in blue and orange, respectively. Gene family gains and losses are indicated in selected nodes: green, novel homology groups (HG); blue, novel core HGs; red, lost HGs. b, Top 14 Protein Class GO hits for novel homology groups (HG) gained across different nodes of chordates, color coded by taxa (legend at the bottom right) and sorted by the Vertebrata node. The largest GO enriched terms are ‘transmembrante signal receptor’ and ‘intercellular signal molecule’ in vertebrates, and ‘defense/immunity protein’ in gnathostomes.
Extended Data Fig. 4 Phylogenetic and retention profile clustering analyses of Hox syntenic regions.
a-d, Bayesian inference phylogenetic trees of amino acid sequences of 4 non-Hox syntenic genes to Hox clusters, Gbx (a), Cbx (b), Hnrnpa (c) and Agap (d), of the inshore hagfish (in red), the sea lamprey (in light blue), the Arctic lamprey (in dark blue) and selected gnathostomes. Orthologs from the European amphioxus Branchiostoma lanceolatum were used as outgroup to root the trees. Posterior probability is indicated in each node. Scales indicate number of substitutions per site. Phylogenetic analyses of Hox genes generally fail to determine orthology due their high conservation and short alignments. The phylogenetic trees of these non-Hox linked genes clearly support the orthology of Hox-α (Gbx, Cbx and Hnrnpa), Hox-δ (Gbx, Hnrnpa and Agap), and Hox-ζ (Hnrnpa) clusters, while β and ε genes always group together, as previously observed for the lamprey17. The alignments used to build the trees, together with the MrBayes parameters and number of generations used to build each tree are provided as Supplementary Files 16–19. e, clustering analysis of retention profiles (see main text) resolved the orthology relationships of Hox-β, Hox-ε, Hox-γ, as well as Hox-ζ clusters. Supported orthologies in each analysis are marked with color-coded rectangles. The location of each cluster is indicated in parenthesis in e (ssc, super scaffold; HiC cl, Hi-C contact cluster, or chromosome). For the clustering analysis of AC1-derived chromosomes in the lamprey and hagfish, we split Hi-C cluster 3 into two halves, each containing one Hox cluster: 3L (coordinates 0–107.78 Mb), 3R (107.78-194 Mb).
Extended Data Fig. 5 Emergence of gnathostome karyotypes via 2R.
a, Chromosomal level phylogenetic trees demonstrate the occurrence of 2R WGD. The two rounds of WGD are color-coded. Cyan and red denote genes encoded by corresponding chicken chromosomes and gar LOCs, respectively. Bootstrap support and posterior probability values of three methods are marked on branches: bootstrap value from RAxML-ng/bootstrap value from IQ-Tree/posterior probability from Astral-III. b, Evolution of gnathostome karyotype through 1R and 2R, with post-1R/pre-2R chromosomal fusions indicated with red lines, and ratios of gene retention asymmetries shown for directly ohnologous chromosomes after 1R and 2R. The size of each ancestral chromosome is proportional to the number of retained genes from ACs.
Extended Data Fig. 6 Contributions of vertebrate ACs to the genomes of hagfish and sea lamprey.
a, Sea lamprey super-scaffolds17 (pseudo-chromosomes) are generally homologous to one single AC except five scaffolds labeled with an arrow. Sea lamprey scaffolds scaf_00001, scaf_00002, scaf_00008, scaf_00010, scaf_00011 and scaf_00023, confounded by missassembly, are not presented here. b, The distribution of the descendant copies of AC genes for the 19 hagfish Hi-C clusters (putative chromosomes). Only significant homologous relationships between hagfish clusters and ACs are shown. Because hagfish cluster 19 is not homologous to any AC, it is presented as a blank block. Genes are color-coded according to its homologous AC.
Extended Data Fig. 7 Overlapping ratio and clustering analysis of ohnologous and orthologous chromosomes.
a, b, AC1 corresponds to five and six mutually paralogous chromosomes in hagfish (a) and sea lamprey (b) genomes, respectively. Numbers in colour-coded cells (bottom left triangle) indicate the OR between two chromosomes. Numbers in white cells (top right triangle) indicate the number of shared retained genes between two chromosomes. Numbers on the diagonal line from top left to bottom right (thick-lined cells) indicate the total number of retained genes of a chromosome. Overlapping rations corresponding to all hagfish chromosomes and lamprey scaffolds are provided in Supplementary File 9. c, Retention profile clustering analysis of gnathostome orthologous chromosomes deriving from AC1. Retained genes are denoted by dark cyan lines. Four orthologous chromosome pairs are defined. d, The clustering found in (c) is the same as that found in the phylogenetic analysis (Extended Data Fig. 5a), demonstrating the reliability of the OR approach.
Extended Data Fig. 8 Fate and cis-regulatory evolution of ohnologs after WGD.
a, b, Gene Ontology enrichment analysis of ohnologs in the hagfish (a) and the chicken (b). Top 20 terms are shown, majority of which are related with development. c, Cumulative distribution of distance of ACRs from the closest TSSs normalized by the average length of intergenic regions in each genome. d, Distribution of the distance from ACRs to closest TSS of developmental ohnologs (Devel.), non-developmental ohnologs (Non-dev.) and non-ohnologous (Non-ohnol.) genes. e, Distribution of the number of ACRs per gene, normalized by the GREAT region length, for developmental ohnologs (Devel.), non-developmental ohnologs (Non-dev.) and non-ohnologous (Non-ohnol.) genes. Sample size for groups in (d) and (e) are identical. n = 143 (Devel.), n = 816 (Non-dev.), n = 7303 (Non-ohnol.). f, Proportion of distal ACRs across different gene functional categories. Within a GREAT-defined region, proximal regulatory sequences were defined as those from 5 kb upstream to 1 kb downstream of a TSS, and the rest of the region was treated as distal. g, Distribution of the number of ACRs of hagfish ohnologs for each category (special., specialization). n = 344 (Redundancy), n = 178 (Subfunction.), n = 334 (Mild special.), n = 240 (Strong special.). h, Distribution of pairwise protein identity for hagfish ohnologous pairs for each category. n = 170 (Redundancy), n = 88 (Subfunction.), n = 165 (Mild special.), n = 120 (Strong special.). i, Distribution of pairwise protein identity for chicken ohnologous pairs for each category. n = 275 (Redundancy), n = 178 (Subfunction.), n = 318 (Mild special.), n = 167 (Strong special.). j, Number of ohnologues with strong specialization in chicken expressed in each tissue. Only the gene in a pair with narrower expression breadth is analyzed. In panels d, e, g-i P values correspond to two-sided Wilcoxon rank sum test between the indicated groups; boxes correspond to the median (centre line) and the first and third quartiles; whiskers extend to the last point no further than 1.5 times the interquartile range from the first and third quartiles. All statistical information for panels d, e, g-i is provided in Supplementary Tables 58–62. Animal illustrations kindly provided by Tamara de Dios Fernández and reproduced with permission from REF. 133.
Supplementary information
Supplementary Information
Supplementary Information.
Supplementary Tables
Supplementary Tables 1–62.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, D., Ren, Y., Uesaka, M. et al. Hagfish genome elucidates vertebrate whole-genome duplication events and their evolutionary consequences. Nat Ecol Evol 8, 519–535 (2024). https://doi.org/10.1038/s41559-023-02299-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41559-023-02299-z
This article is cited by
-
A jawless-fish genome untangles the history of vertebrate genome multiplications
Nature (2024)
-
Extensive DNA methylome rearrangement during early lamprey embryogenesis
Nature Communications (2024)
